Esta prueba ofrece detección molecular mediante secuenciación de próxima generación (NGS) de variantes para trastornos autosómicos recesivos y ligados al cromosoma X específicos y permite la detección de portadores de individuos independientemente de su ascendencia u origen geográfico. La detección de portadores tiene como objetivo identificar a las parejas que tienen un mayor riesgo de tener un hijo afectado para facilitar la toma de decisiones reproductivas informadas; Este gran panel puede ser particularmente útil en el entorno previo a la concepción o de fertilidad. Como se trata de una prueba de detección, este panel portador no está diseñado para utilizarse con fines de diagnóstico. Si desea realizar pruebas genéticas de diagnóstico, llame a Servicios al cliente de Genomic (GENEINFO) al 866.436.3463 para analizar el caso con un asesor genético de Quest. Esta prueba analiza genes asociados con 421 condiciones, incluidas las 150 condiciones incluidas en la pantalla de portadores QHerit™ Extended. Las condiciones incluidas en este panel incluyen: deficiencia de 17-beta-hidroxiesteroide deshidrogenasa, tipo III (HSD17B3); deficiencia de 3-beta-hidroxiesteroide deshidrogenasa, tipo II (HSD3B2); deficiencia de 3-hidroxi-3-metilglutaril-coA liasa (HMGCL); deficiencia de 3-hidroxiacil-CoA deshidrogenasa (HADH); deficiencia de 3-metilcrotonil-CoA carboxilasa 1 (MCCC1); deficiencia de 3-metilcrotonil-CoA carboxilasa 2 (MCCC2); aciduria 3-metilglutacónica tipo III/síndrome de Costeff (OPA3); deficiencia de 6-piruvoil-tetrahidropterina sintasa (PTS); Abetalipoproteinemia (MTTP); Acromatopsia relacionada con CNGB3 (CNGB3); Acrodermatitis enteropática (SLC39A4); Síndrome de insuficiencia renal mioclónica de acción (SCARB2); Insuficiencia hepática infantil aguda (TRMU); Deficiencia de adenosina desaminasa (ADA); Adrenoleucodistrofia ligada al cromosoma X (ABCD1); Agenesia del cuerpo calloso con neuropatía periférica (SLC12A6); síndrome de Aicardi-Goutières 2 (RNASEH2B); síndrome de Aicardi-Goutières 3 (RNASEH2C); síndrome de Aicardi-Goutières 4 (RNASEH2A); síndrome de Aicardi-Goutières 5 (SAMHD1); Deficiencia de alfa-1 antitripsina (SERPINA1); Alfamanosidosis (MAN2B1); Alfa-talasemia (HBA1/HBA2); síndrome de discapacidad intelectual alfa-talasemia ligado al cromosoma X (ATRX); síndrome de Alport, relacionado con COL4A3 (COL4A3); síndrome de Alport, relacionado con COL4A4 (COL4A4); Síndrome de Alport, relacionado con COL4A5, ligado al cromosoma X (COL4A5); Síndrome de Alstrom (ALMS1); síndrome de epilepsia infantil amish (ST3GAL5); Argininemia (ARG1); Aciduria argininosuccínica (ASL); Deficiencia de aromatasa (CYP19A1); Artrogriposis, retraso mental y convulsiones (SLC35A3); Deficiencia de asparagina sintetasa (ASNS); Aspartilglucosaminuria (AGA); Ataxia con deficiencia aislada de vitamina E (TTPA); Ataxia-telangiectasia (ATM); Trastorno similar a ataxia-telangiectasia 1 (MRE11); Síndrome poliglandular autoinmune tipo 1 (AIRE); Ictiosis congénita autosómica recesiva 1 (TGM1); Enfermedad renal poliquística autosómica recesiva (PKHD1); síndrome de Bardet-Biedl 1 (BBS1); síndrome de Bardet-Biedl 10 (BBS10); síndrome de Bardet-Biedl 12 (BBS12); síndrome de Bardet-Biedl 2 (BBS2); síndrome de Bardet-Biedl 4 (BBS4); síndrome de Bardet-Biedl 6 (MKKS); síndrome de Bardet-Biedl 7 (BBS7); síndrome de Bardet-Biedl 8 (TTC8); síndrome de Bardet-Biedl 9 (BBS9); Síndrome de linfocitos desnudos, tipo II (CIITA); síndrome de Barth (TAFAZZIN) ; síndrome de Bartter, tipo 4A (BSND); síndrome de Bernard-Soulier, tipo A (GP1BA); síndrome de Bernard-Soulier, tipo C (GP9); Beta hemoglobinopatías (HBB); Deficiencia de beta-cetotiolasa (ACAT1); Deficiencia de beta-ureidopropionasa (UPB1); Polimicrogiria frontoparietal bilateral (ADGRG1); Deficiencia de biotinidasa (BTD); síndrome de Bloom (BLM); enfermedad de Canavan (ASPA); Deficiencia de carbamoil fosfato sintetasa I (CPS1); deficiencia de carnitina, primario sistémico (SLC22A5); Deficiencia de carnitina palmitoiltransferasa I (CPT1A); Deficiencia de carnitina palmitoiltransferasa II (CPT2); Deficiencia de carnitina-acilcarnitina translocasa (SLC25A20); síndrome de Carpenter (RAB23); Hipoplasia cartílago-pelo (RMRP); Condiciones relacionadas con CEP290 (CEP290); síndrome cerebrooculofacioesquelético 1 / síndrome de Cockayne, tipo B (ERCC6); xantomatosis cerebrotendinosa (CYP27A1); enfermedad de Charcot-Marie-Tooth, tipo 1X (GJB1); enfermedad de Charcot-Marie-Tooth, tipo 4D (NDRG1); síndrome de Chediak-Higashi (LYST); Colestasis intrahepática familiar progresiva 4 (TJP2); Coreoacantocitosis (VPS13A); coroideremia ligada al cromosoma X (CHM); Enfermedad granulomatosa crónica 4 (CYBA); Enfermedad granulomatosa crónica ligada al cromosoma X (CYBB); Ciliopatías relacionadas con RPGRIP1L (RPGRIP1L); Deficiencia de citrina / Citrulinemia tipo II (SLC25A13); Citrulinemia tipo I (ASS1); síndrome de Cockayne, tipo A (ERCC8); síndrome de Cohen (VPS13B); Aciduria combinada malónica y metilmalónica (ACSF3); Aciduria metilmalónica y homocistinuria combinadas, tipo cblC/deficiencia de cobalamina C (MMACHC); Aciduria metilmalónica y homocistinuria combinadas, tipo cblD/deficiencia de cobalamina D (MMADHC); Deficiencia combinada de fosforilación oxidativa 1 (GFM1); Deficiencia combinada de fosforilación oxidativa 3 (TSFM); Deficiencia combinada de fosforilación oxidativa 6 (AIFM1); Deficiencia combinada de hormona pituitaria 3 (LHX3); Deficiencia combinada de hormonas pituitarias, tipo 2 (PROP1); Hiperplasia suprarrenal congénita (CAH) debida a deficiencia de 11-beta-hidroxilasa (CYP11B1); Hiperplasia suprarrenal congénita (CAH) debida a deficiencia de 17-alfa-hidroxilasa (CYP17A1); Hiperplasia suprarrenal congénita (CAH) debida a deficiencia de 21-hidroxilasa (CYP21A2); Trombocitopenia amegacariocítica congénita (MPL); Trastorno congénito de la glicosilación, tipo Ia (PMM2); Trastorno congénito de la glicosilación, tipo Ib (MPI); Trastorno congénito de la glicosilación, tipo Ic (ALG6); Ictiosis congénita relacionada con ABCA12 (ABCA12); Insensibilidad congénita al dolor con anhidrosis (NTRK1); Distrofia-distroglicanopatía muscular congénita 1 (POMT1); Síndrome miasténico congénito relacionado con CHAT (CHAT); Síndrome miasténico congénito relacionado con CHRNE (CHRNE); Síndrome miasténico congénito relacionado con DOK7 (DOK7); Síndrome miasténico congénito relacionado con RAPSN (RAPSN); Neutropenia congénita relacionada con HAX1 (HAX1); Síndrome de distrofia corneal y sordera perceptiva (SLC4A11); Deficiencia de corticosterona metiloxidasa (CYP11B2); distrofias retinianas relacionadas con CRB1 (CRB1); Defecto del transportador de creatina, relacionado con SLC6A8, ligado al cromosoma X/síndrome de deficiencia de creatina cerebral (SLC6A8); Fibrosis quística (CFTR); Cistinosis (CTNS); Deficiencia de proteína D-bifuncional (HSD17B4); Trastornos relacionados con DCX (DCX); Enfermedad de Dent (CLCN5); displasia desbuquois tipo I (CANT1); Deficiencia de dihidrolipoamida deshidrogenasa (DLD); Deficiencia de dihidropirimidina deshidrogenasa (DPYD); Distrofia muscular de Duchenne/Becker, ligado al cromosoma X (DMD); Disqueratosis congénita relacionada con RTEL1 (RTEL1); Disqueratosis congénita ligada al cromosoma X (DKC1); Epidermólisis ampollosa distrófica, relacionada con COL7A1 (COL7A1); síndrome de Ehlers-Danlos, tipo dermatosparaxis (ADAMTS2); síndrome de Ellis-van Creveld (EVC); síndrome de Ellis-van Creveld (EVC2); distrofia muscular de Emery-Dreifuss ligada al cromosoma X (EMD); Síndrome del cono S mejorado (NR2E3); Condiciones relacionadas con ERCC2 (ERCC2); encefalopatía etilmalónica (ETHE1); enfermedad de Fabry ligada al cromosoma X (GLA); Deficiencia de factor IX/Hemofilia B (F9); Deficiencia de factor XI/Hemofilia C (F11); Disautonomía familiar (ELP1); Linfohistiocitosis hemofagocítica familiar 2 (PRF1); Linfohistiocitosis hemofagocítica familiar 4 (STX11); Linfohistiocitosis hemofagocítica familiar 5 (STXBP2); Hipercolesterolemia familiar relacionada con LDLRAP1 (LDLRAP1); Hipercolesterolemia familiar relacionada con LDLR (LDLR); Hiperinsulinismo familiar relacionado con KCNJ11 (KCNJ11); Hiperinsulinismo familiar relacionado con ABAyuno de 8 a 12 horas antes del examen. Seguir las indicaciones médicas específicas para la prueba.0
El nuevo arte de cuidar tu salud divirtiendote Si últimamente has sentido que el plan de desvelarte y sacrificar tu mañana siguiente ya no va contigo, no eres el único. Actualmente, la forma de socializar dio un giro de 180°. El nuevo lujo no es salir de noche; es despertar con energía, disfrutar de un ... ¿Qué es una Coffee Party? Descubre la tendencia más saludable del 2026
Why Nuevo Polanco Medical Center is Your Best Choice? Are you looking for premium dental care but worried about the skyrocketing costs in the U.S.? Welcome to Nuevo Polanco Medical Center (NPMC) in Mexico City, your #1 destination for world-class dental health services. At NPMC, we combine quality, affordability, and efficiency in one luxurious place, ... Premium Dental Care in Mexico City:
Introducción: La Importancia del Monitoreo Preventivo La salud cardiovascular es el pilar de una vida larga y plena. Realizar chequeos preventivos de manera regular es la mejor estrategia para detectar silenciosamente los factores de riesgo antes de que se conviertan en problemas graves. Este paquete de chequeo integral está diseñado para ofrecer una visión profunda ... Paquete de Chequeo de Salud Cardiovascular Integral Un Estudio para tu Corazón y Bienestar General
Los genes BRCA1 y BRCA2 son componentes críticos en el mantenimiento de la salud celular. Su función principal es actuar como “guardianes del genoma”, reparando el material genético (ADN) dañado y asegurando que las células se dividan de manera controlada y precisa.Cuando estos genes presentan una mutación hereditaria una alteración que compromete su función la ... Un enfoque Preventivo: Genes BRCA1 y BRCA2
La fibrosis quística (FQ) es una enfermedad genética crónica y multisistémica que afectaprincipalmente a los pulmones y al sistema digestivo, aunque también puede comprometerel hígado, el páncreas, los riñones y el aparato reproductor. Se produce por mutaciones enel gen CFTR, responsable de codificar una proteína esencial para el transporte de cloro ybicarbonato en las células ... FIBRÓSIS QUÍSTICA: Una Revisión científica y clínica.



 El nuevo arte de cuidar tu salud divirtiendote Si últimamente has sentido que el plan de desvelarte y sacrificar tu mañana siguiente ya no va contigo, no eres el único. Actualmente, la forma de socializar dio un giro de 180°. El nuevo lujo no es salir de noche; es despertar con energía, disfrutar de un ... ¿Qué es una Coffee Party? Descubre la tendencia más saludable del 2026
El nuevo arte de cuidar tu salud divirtiendote Si últimamente has sentido que el plan de desvelarte y sacrificar tu mañana siguiente ya no va contigo, no eres el único. Actualmente, la forma de socializar dio un giro de 180°. El nuevo lujo no es salir de noche; es despertar con energía, disfrutar de un ... ¿Qué es una Coffee Party? Descubre la tendencia más saludable del 2026 Why Nuevo Polanco Medical Center is Your Best Choice? Are you looking for premium dental care but worried about the skyrocketing costs in the U.S.? Welcome to Nuevo Polanco Medical Center (NPMC) in Mexico City, your #1 destination for world-class dental health services. At NPMC, we combine quality, affordability, and efficiency in one luxurious place, ... Premium Dental Care in Mexico City:
Why Nuevo Polanco Medical Center is Your Best Choice? Are you looking for premium dental care but worried about the skyrocketing costs in the U.S.? Welcome to Nuevo Polanco Medical Center (NPMC) in Mexico City, your #1 destination for world-class dental health services. At NPMC, we combine quality, affordability, and efficiency in one luxurious place, ... Premium Dental Care in Mexico City: Introducción: La Importancia del Monitoreo Preventivo La salud cardiovascular es el pilar de una vida larga y plena. Realizar chequeos preventivos de manera regular es la mejor estrategia para detectar silenciosamente los factores de riesgo antes de que se conviertan en problemas graves. Este paquete de chequeo integral está diseñado para ofrecer una visión profunda ... Paquete de Chequeo de Salud Cardiovascular Integral Un Estudio para tu Corazón y Bienestar General
Introducción: La Importancia del Monitoreo Preventivo La salud cardiovascular es el pilar de una vida larga y plena. Realizar chequeos preventivos de manera regular es la mejor estrategia para detectar silenciosamente los factores de riesgo antes de que se conviertan en problemas graves. Este paquete de chequeo integral está diseñado para ofrecer una visión profunda ... Paquete de Chequeo de Salud Cardiovascular Integral Un Estudio para tu Corazón y Bienestar General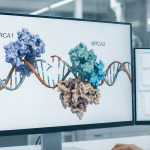 Los genes BRCA1 y BRCA2 son componentes críticos en el mantenimiento de la salud celular. Su función principal es actuar como “guardianes del genoma”, reparando el material genético (ADN) dañado y asegurando que las células se dividan de manera controlada y precisa.Cuando estos genes presentan una mutación hereditaria una alteración que compromete su función la ... Un enfoque Preventivo: Genes BRCA1 y BRCA2
Los genes BRCA1 y BRCA2 son componentes críticos en el mantenimiento de la salud celular. Su función principal es actuar como “guardianes del genoma”, reparando el material genético (ADN) dañado y asegurando que las células se dividan de manera controlada y precisa.Cuando estos genes presentan una mutación hereditaria una alteración que compromete su función la ... Un enfoque Preventivo: Genes BRCA1 y BRCA2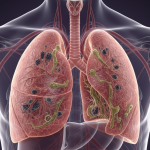 La fibrosis quística (FQ) es una enfermedad genética crónica y multisistémica que afectaprincipalmente a los pulmones y al sistema digestivo, aunque también puede comprometerel hígado, el páncreas, los riñones y el aparato reproductor. Se produce por mutaciones enel gen CFTR, responsable de codificar una proteína esencial para el transporte de cloro ybicarbonato en las células ... FIBRÓSIS QUÍSTICA: Una Revisión científica y clínica.
La fibrosis quística (FQ) es una enfermedad genética crónica y multisistémica que afectaprincipalmente a los pulmones y al sistema digestivo, aunque también puede comprometerel hígado, el páncreas, los riñones y el aparato reproductor. Se produce por mutaciones enel gen CFTR, responsable de codificar una proteína esencial para el transporte de cloro ybicarbonato en las células ... FIBRÓSIS QUÍSTICA: Una Revisión científica y clínica.