Es un panel de 161 genes que incluye la evaluación de variantes no codificantes. Además, también incluye el genoma mitocondrial heredado por la madre. Es ideal para pacientes con miopatía distal o sospecha clínica de distrofia muscular. Incluye el Panel de Miopatía Nemalínica más pequeño, el panel de LGMD y distrofia muscular congénita, el panel de distrofia muscular de Emery-Dreifuss y el panel de trastornos relacionados con el colágeno tipo VI. Las distrofias musculares y las miopatías son un grupo complejo de trastornos neuromusculares o musculoesqueléticos que típicamente resultan en debilidad muscular progresiva. La edad de aparición, los grupos musculares afectados y los síntomas adicionales dependen del tipo de enfermedad. La distrofia muscular de la cintura escapular (LGMD, por sus siglas en inglés) es un grupo de trastornos con atrofia y debilidad de los músculos proximales de la cintura escapular, que normalmente no afecta al corazón ni a los músculos bulbares. Sin embargo, se pueden observar alteraciones cardíacas y respiratorias en ciertas formas de LGMD. En la distrofia muscular congénita (CMD), el inicio de la debilidad muscular generalmente se presenta en el período neonatal o en la primera infancia. La gravedad clínica, la edad de inicio y la progresión de la enfermedad son muy variables entre las diferentes formas de LGMD/CMD. Los fenotipos se superponen tanto dentro de los subtipos de CMD como entre las distrofias musculares congénitas, las miopatías congénitas y las LGMD. La distrofia muscular de Emery-Dreifuss (EDMD) es una afección que afecta principalmente al músculo esquelético y al corazón. Por lo general, se presenta en la primera infancia con contracturas, que restringen el movimiento de ciertas articulaciones, con mayor frecuencia codos, tobillos, y cuello. La mayoría de los pacientes también experimentan debilidad y atrofia muscular lentamente progresiva, comenzando con los músculos de la parte superior del brazo y la parte inferior de la pierna y progresando hacia los hombros y las caderas. Casi todos los pacientes con EDMD tienen afectación cardíaca en la edad adulta. Se presenta clínicamente como defectos de conducción cardiaca y/o arritmias. El fenotipo de miocardiopatía generalmente se clasifica como miocardiopatía dilatada (DCM), pero también se han descrito ARVC y miocardiopatías hipertróficas (MCH). Una pequeña proporción de pacientes con EDMD autosómica dominante experimenta manifestaciones cardíacas sin debilidad o desgaste del músculo esquelético. Casi todos los pacientes con EDMD tienen afectación cardíaca en la edad adulta. Se presenta clínicamente como defectos de conducción cardiaca y/o arritmias. El fenotipo de miocardiopatía generalmente se clasifica como miocardiopatía dilatada (DCM), pero también se han descrito ARVC y miocardiopatías hipertróficas (MCH). Una pequeña proporción de pacientes con EDMD autosómica dominante experimenta manifestaciones cardíacas sin debilidad o desgaste del músculo esquelético. Casi todos los pacientes con EDMD tienen afectación cardíaca en la edad adulta. Se presenta clínicamente como defectos de conducción cardiaca y/o arritmias. El fenotipo de miocardiopatía generalmente se clasifica como miocardiopatía dilatada (DCM), pero también se han descrito ARVC y miocardiopatías hipertróficas (MCH). Una pequeña proporción de pacientes con EDMD autosómica dominante experimenta manifestaciones cardíacas sin debilidad o desgaste del músculo esquelético. Las distrofinopatías incluyen un espectro de enfermedades musculares que van desde asintomáticas con un aumento en la concentración sérica de creatina fosfocinasa hasta las enfermedades musculares progresivas graves que se clasifican como distrofia muscular de Duchenne o Becker (DMD o BMD) cuando el músculo esquelético es el principal afectado y asociado a DMD. DCM cuando el corazón se ve afectado principalmente. La DMD generalmente se presenta en la primera infancia con hitos retrasados, incluidos retrasos para sentarse y pararse de forma independiente. La DMD es rápidamente progresiva, y los niños afectados dependen de una silla de ruedas a la edad de 13 años y la miocardiopatía se presenta poco después. La BMD se caracteriza por una debilidad del músculo esquelético de aparición tardía; algunos individuos permanecen ambulatorios hasta los 20 años. Sin embargo, la insuficiencia cardíaca por DCM es una causa común de muerte a mediados de los 40 años. La DCM asociada a DMD se caracteriza por dilatación del ventrículo izquierdo e insuficiencia cardíaca congestiva. Hembras heterocigotas para una variante patógena enDMD tienen un mayor riesgo de DCM. Los trastornos relacionados con el colágeno tipo VI ahora se reconocen como un continuo de fenotipos superpuestos con miopatía de Bethlem en el extremo leve y distrofia muscular congénita de Ullrich (UCMD) en el extremo grave. Entre estos fenotipos, hay distrofia muscular de cinturas y miopatía miosclerótica relacionada con el colágeno tipo VI. La miopatía de Bethlem se caracteriza por debilidad proximal y contracturas variables. Los codos, los tobillos y los dedos son los más afectados. Si el inicio es en la primera infancia, se evidencian retraso en los hitos motores, debilidad muscular y contracturas. Los pacientes con inicio en la edad adulta eventualmente requieren apoyo ambulatorio. La UCMD se caracteriza por debilidad muscular congénita, contracturas de las articulaciones proximales e hiperlaxitud notable de las articulaciones distales. Los niños afectados rara vez adquieren la capacidad de caminar de forma independiente y desarrollan rigidez de la columna y escoliosis. La insuficiencia respiratoria es una causa común de muerte en la primera y segunda década de la vida. La inteligencia es normal tanto en la miopatía de Bethlem como en la UCMD. La miopatía nemalínica se caracteriza por debilidad, hipotonía y reflejos tendinosos profundos disminuidos o ausentes. Histopatológicamente, los cuerpos de nemalina se detectan en la biopsia muscular. La clasificación clínica define seis formas de miopatía nemalínica, que se clasifican según el inicio y la gravedad de la afectación motora y respiratoria. Se produce una superposición considerable entre las formas.Ayuno de 8 a 12 horas antes del examen. Seguir las indicaciones médicas específicas para la prueba.0
Why Nuevo Polanco Medical Center is Your Best Choice? Are you looking for premium dental care but worried about the skyrocketing costs in the U.S.? Welcome to Nuevo Polanco Medical Center (NPMC) in Mexico City, your #1 destination for world-class dental health services. At NPMC, we combine quality, affordability, and efficiency in one luxurious place, ... Premium Dental Care in Mexico City:
Introducción: La Importancia del Monitoreo Preventivo La salud cardiovascular es el pilar de una vida larga y plena. Realizar chequeos preventivos de manera regular es la mejor estrategia para detectar silenciosamente los factores de riesgo antes de que se conviertan en problemas graves. Este paquete de chequeo integral está diseñado para ofrecer una visión profunda ... Paquete de Chequeo de Salud Cardiovascular Integral Un Estudio para tu Corazón y Bienestar General
Los genes BRCA1 y BRCA2 son componentes críticos en el mantenimiento de la salud celular. Su función principal es actuar como “guardianes del genoma”, reparando el material genético (ADN) dañado y asegurando que las células se dividan de manera controlada y precisa.Cuando estos genes presentan una mutación hereditaria una alteración que compromete su función la ... Un enfoque Preventivo: Genes BRCA1 y BRCA2
La fibrosis quística (FQ) es una enfermedad genética crónica y multisistémica que afectaprincipalmente a los pulmones y al sistema digestivo, aunque también puede comprometerel hígado, el páncreas, los riñones y el aparato reproductor. Se produce por mutaciones enel gen CFTR, responsable de codificar una proteína esencial para el transporte de cloro ybicarbonato en las células ... FIBRÓSIS QUÍSTICA: Una Revisión científica y clínica.
¿Sabías que más del 70% de las enfermedades crónicas podrían prevenirse con un diagnóstico oportuno? La ciencia médica ha dado un salto exponencial en la detección temprana de enfermedades, permitiendo conocer el estado de salud con estudios asequibles y rápidos. Gracias a los avances en estudios de imagenología, genética y laboratorio clínico, hoy es posible ... Descubre el Futuro de la Salud Preventiva: Cómo la Tecnología y la Genética Pueden Salvar Vidas



 Why Nuevo Polanco Medical Center is Your Best Choice? Are you looking for premium dental care but worried about the skyrocketing costs in the U.S.? Welcome to Nuevo Polanco Medical Center (NPMC) in Mexico City, your #1 destination for world-class dental health services. At NPMC, we combine quality, affordability, and efficiency in one luxurious place, ... Premium Dental Care in Mexico City:
Why Nuevo Polanco Medical Center is Your Best Choice? Are you looking for premium dental care but worried about the skyrocketing costs in the U.S.? Welcome to Nuevo Polanco Medical Center (NPMC) in Mexico City, your #1 destination for world-class dental health services. At NPMC, we combine quality, affordability, and efficiency in one luxurious place, ... Premium Dental Care in Mexico City: Introducción: La Importancia del Monitoreo Preventivo La salud cardiovascular es el pilar de una vida larga y plena. Realizar chequeos preventivos de manera regular es la mejor estrategia para detectar silenciosamente los factores de riesgo antes de que se conviertan en problemas graves. Este paquete de chequeo integral está diseñado para ofrecer una visión profunda ... Paquete de Chequeo de Salud Cardiovascular Integral Un Estudio para tu Corazón y Bienestar General
Introducción: La Importancia del Monitoreo Preventivo La salud cardiovascular es el pilar de una vida larga y plena. Realizar chequeos preventivos de manera regular es la mejor estrategia para detectar silenciosamente los factores de riesgo antes de que se conviertan en problemas graves. Este paquete de chequeo integral está diseñado para ofrecer una visión profunda ... Paquete de Chequeo de Salud Cardiovascular Integral Un Estudio para tu Corazón y Bienestar General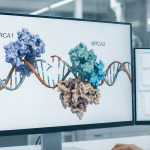 Los genes BRCA1 y BRCA2 son componentes críticos en el mantenimiento de la salud celular. Su función principal es actuar como “guardianes del genoma”, reparando el material genético (ADN) dañado y asegurando que las células se dividan de manera controlada y precisa.Cuando estos genes presentan una mutación hereditaria una alteración que compromete su función la ... Un enfoque Preventivo: Genes BRCA1 y BRCA2
Los genes BRCA1 y BRCA2 son componentes críticos en el mantenimiento de la salud celular. Su función principal es actuar como “guardianes del genoma”, reparando el material genético (ADN) dañado y asegurando que las células se dividan de manera controlada y precisa.Cuando estos genes presentan una mutación hereditaria una alteración que compromete su función la ... Un enfoque Preventivo: Genes BRCA1 y BRCA2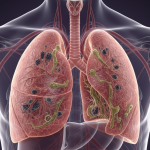 La fibrosis quística (FQ) es una enfermedad genética crónica y multisistémica que afectaprincipalmente a los pulmones y al sistema digestivo, aunque también puede comprometerel hígado, el páncreas, los riñones y el aparato reproductor. Se produce por mutaciones enel gen CFTR, responsable de codificar una proteína esencial para el transporte de cloro ybicarbonato en las células ... FIBRÓSIS QUÍSTICA: Una Revisión científica y clínica.
La fibrosis quística (FQ) es una enfermedad genética crónica y multisistémica que afectaprincipalmente a los pulmones y al sistema digestivo, aunque también puede comprometerel hígado, el páncreas, los riñones y el aparato reproductor. Se produce por mutaciones enel gen CFTR, responsable de codificar una proteína esencial para el transporte de cloro ybicarbonato en las células ... FIBRÓSIS QUÍSTICA: Una Revisión científica y clínica. ¿Sabías que más del 70% de las enfermedades crónicas podrían prevenirse con un diagnóstico oportuno? La ciencia médica ha dado un salto exponencial en la detección temprana de enfermedades, permitiendo conocer el estado de salud con estudios asequibles y rápidos. Gracias a los avances en estudios de imagenología, genética y laboratorio clínico, hoy es posible ... Descubre el Futuro de la Salud Preventiva: Cómo la Tecnología y la Genética Pueden Salvar Vidas
¿Sabías que más del 70% de las enfermedades crónicas podrían prevenirse con un diagnóstico oportuno? La ciencia médica ha dado un salto exponencial en la detección temprana de enfermedades, permitiendo conocer el estado de salud con estudios asequibles y rápidos. Gracias a los avances en estudios de imagenología, genética y laboratorio clínico, hoy es posible ... Descubre el Futuro de la Salud Preventiva: Cómo la Tecnología y la Genética Pueden Salvar Vidas